
Speciale Natale 2022
Con il vostro contributo si può sostenere la ricerca, unico mezzo per dare sempre maggioresperanza a chi, più sfortunato di […]
Con il vostro contributo si può sostenere la ricerca, unico mezzo per dare sempre maggioresperanza a chi, più sfortunato di […]

Domenica 2 Ottobre parteciperemo alla Festa del Volontario nella splendida cornice di Prato della Valle a Padova. Vi aspettiamo!

Quando abbiamo ricevuto la richiesta da parte dell’equipe del Prof. Livio Trentin, del dipartimento di Ematologia e Immunologia Clinica, di
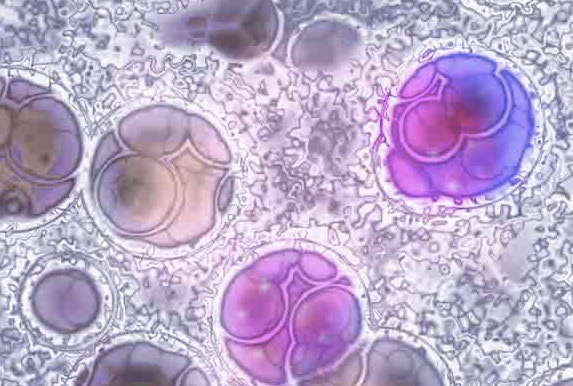
RCV “Ricerca Per Credere Nella Vita” • ODV finanzia il progetto con 55.000,00 euro. Il progetto si propone di proseguire

Il 5 x mille è una quota di imposte, a cui lo Stato rinuncia per destinarla alle organizzazioni no-profit per

L’assemblea dei soci riunitasi giovedì 28 aprile 2022 ha eletto le nuove cariche sociali. Nomina Direttivo L’attuale Presidente Ing. Renzo

QUOTE MINIME DI ISCRIZIONE: Le adesioni verranno raccolte dalla segreteria del Circolo. Per informazioni: 049 880 54 94
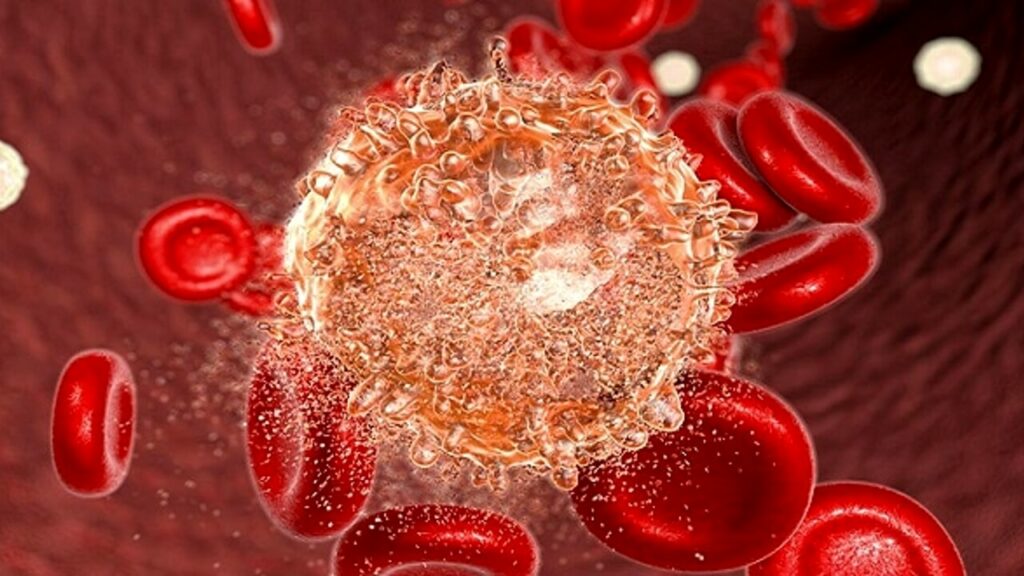
Un team di ricercatori padovani ha scoperto come la leucemia evolve in sindrome di Richter e come ottimizzare l’uso dei

Con il vostro contributo si può sostenere la Ricerca, unico mezzo per dare sempre maggiore speranza a chi, più sfortunato
